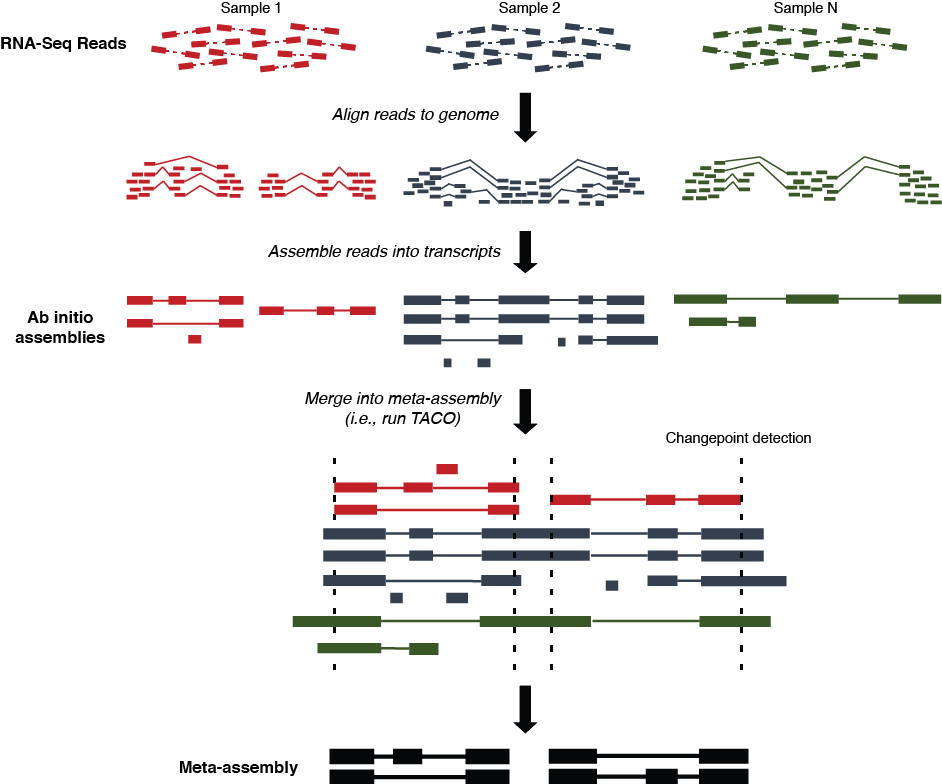
Multi-sample transcriptome assembly protocol
- Sequence reads from each dataset are aligned to the reference genome using
STAR,
HISAT2, or another RNA-Seq aligner
- Read alignments (SAM/BAM/CRAM format) from each dataset are assembled into transfrags using
Cufflinks,
StringTie, or another ab initio transcriptome assembly tool.
- Multi-sample assembly is performed on the collection of
ab initio assemblies (GTF format) by
TACO, resulting in a consensus merged transcriptome
- Downstream analysis of gene expression, conservation, variation, and functionality can be performed
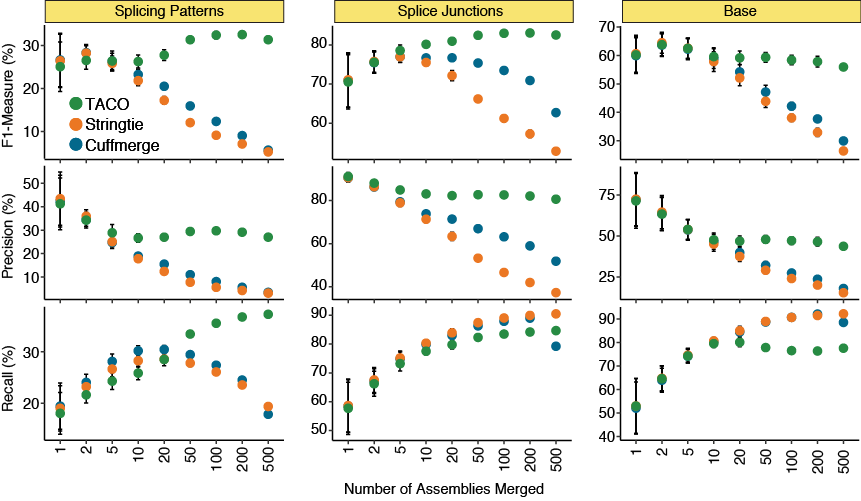
Varying the number of input assemblies
Performance metrics for TACO, Cuffmerge, and StringTie when merging different numbers of input assemblies. Recall (i.e., sensitivity), precision and the F-measure for all three tools were assessed for splicing patterns, splice junctions, and bases. Twenty
batches were randomly selected at each number of assemblies. Points represent the mean statistic across the 20 runs, error bars represent the 95% confidence interval.
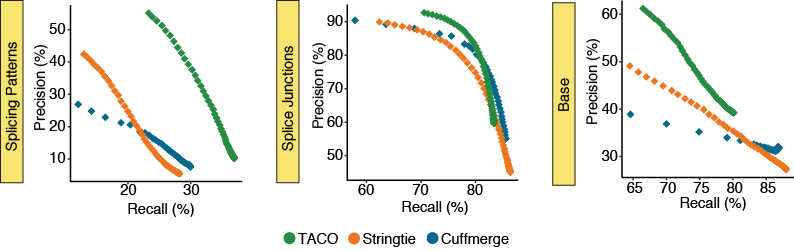
Varying isoform fraction cutoff
The parameter --isoform-frac FRAC configures TACO to only report isoforms with abundance ratio more than
FRAC of the most abundant transcript in the locus
The precision-recall (PR) plots depict the performance for the three merging tools at 50 different isoform fraction cutoffs ranging from 0.001-0.999. Statistics describe the merger of the 55 breast cancer cell lines in the CCLE.
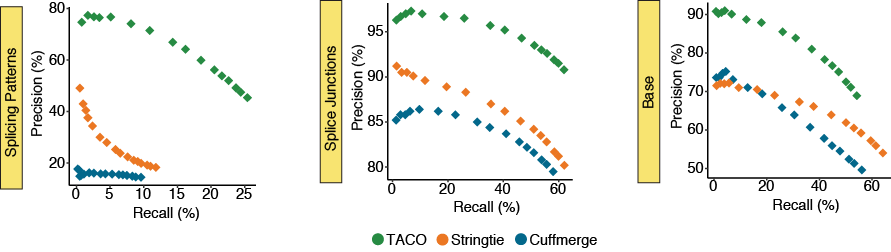
Performance for highly expressed transcripts
The precision-recall (PR) plots depict the performance for the three merging tools across subsets of the top 500, 1k, 1.5k, 2k, 3k, 5k, 7k, 10k, 12k, 15k, 18k, 20k, 22k, 25k, 27k, and 30k highest expressed transcripts in the merger
of 55 breast cancer cell lines from the CCLE
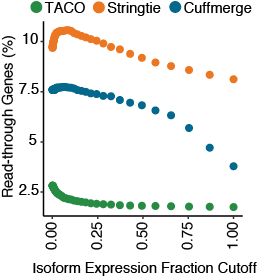
Fraction of read-through transcripts
Scatterplot depicting the fraction of read-through genes produced in the multi-sample assembly from the three merge tools. Gene designated as being a read-through if it contains at least one transcript isoform bearing exonic overlap
with two separate protein-coding genes on the same strand. Fraction of read-throughs was determined for assemblies at 50 different isoform fraction cutoffs ranging from 0.001-0.999.
Releases
| Version |
Date |
Changes |
Execuatble Binaries |
Source Package |
| v0.7.3 |
9/25/2017 |
Bug fix permitting spaces in GTF attributes |
Mac
Linux
|
Source |
| v0.7.2 |
5/15/2017 |
Added option to supply additional splice motifs to permit in splice junction filter |
Mac
Linux
|
Source |
| v0.7.0 |
2/13/2017 |
Fixed bug in refcomp code for annotation of intragenic overlap |
Mac
Linux
|
Source |
| v0.6.3 |
2/01/2017 |
Cleaner Input Instructions |
Mac
Linux
|
Source |
| v0.6.2 |
1/31/2017 |
Refcomp Optimized |
Mac
Linux
|
Source |
| v0.6.1 |
1/28/2017 |
Added Refcomp Source Code and Added Mac Support |
Mac
Linux
|
Source |
| v0.6.0 |
12/13/2016 |
Added Reference Comparison Tool |
Mac
Linux
|
Source |
| v0.5.1 |
4/28/2016 |
Version used for manuscript submission |
Mac
Linux
|
Source |
Source Code
The TACO GitHub repository is located
here.
Build Code
More information on the build procedure is located here.
Test Installation
Instructions on testing the installation are here.
For your convenience we are providing links for human references.
Be sure to gunzip the file before running with TACO or TACO-refcomp.
Unpack the downloaded binary package:
$ tar -xpvfz taco-<RELEASE>.tar.gz
On some machines, it may be necessary to change the permissions of the executable
$ cd taco-<RELEASE>/
$ chmod +x taco_run
To test installatoin, download the following two test data files and place them in the same directory as the TACO executable:
In the directory where you placed the two downloaded files and unpacked the binary file, type:
./taco_run --gtf-expr-attr expr taco_test_files.txt
Note:
Our sample dataset has already been processed by TACO, which converts
the attribute specified by ‑‑gtf-expr-attr (e.g. FPKM)
into expr. Hence, we use the argument
‑‑gtf-expr-attr expr here.
You should see the following output:
2016-03-30 14:54:02,097 pid=89792 INFO - taco version 0.5.0
2016-03-30 14:54:02,097 pid=89792 INFO - ------------------------------------------------------------------------------
2016-03-30 14:54:02,097 pid=89792 INFO - verbose logging: False
2016-03-30 14:54:02,097 pid=89792 INFO - num processes: 1
2016-03-30 14:54:02,097 pid=89792 INFO - output directory: taco
2016-03-30 14:54:02,097 pid=89792 INFO - min transfrag length: 200
2016-03-30 14:54:02,098 pid=89792 INFO - min expression: 0.5
2016-03-30 14:54:02,098 pid=89792 INFO - reference GTF file: None
2016-03-30 14:54:02,098 pid=89792 INFO - guided assembly mode: False
2016-03-30 14:54:02,098 pid=89792 INFO - guided strand mode: False
2016-03-30 14:54:02,098 pid=89792 INFO - guided ends mode: False
2016-03-30 14:54:02,098 pid=89792 INFO - GTF expression attribute: expr
2016-03-30 14:54:02,098 pid=89792 INFO - isoform fraction: 0.05
2016-03-30 14:54:02,098 pid=89792 INFO - max_isoforms: 0
2016-03-30 14:54:02,098 pid=89792 INFO - change point: True
2016-03-30 14:54:02,098 pid=89792 INFO - change point pvalue: 0.001
2016-03-30 14:54:02,098 pid=89792 INFO - change point fold change: 0.85
2016-03-30 14:54:02,098 pid=89792 INFO - change point trim: True
2016-03-30 14:54:02,098 pid=89792 INFO - path graph loss threshold: 0.1
2016-03-30 14:54:02,098 pid=89792 INFO - path frac: 0.0
2016-03-30 14:54:02,099 pid=89792 INFO - max paths: 0
2016-03-30 14:54:02,099 pid=89792 INFO - Samples: 1
2016-03-30 14:54:02,099 pid=89792 INFO - Aggregating GTF files
2016-03-30 14:54:02,210 pid=89792 INFO - Sorting GTF
2016-03-30 14:54:02,236 pid=89792 INFO - Indexing Loci
2016-03-30 14:54:02,239 pid=89792 INFO - Assembling GTF files
2016-03-30 14:54:02,239 pid=89792 INFO - Assembling in parallel using 1 processes
2016-03-30 14:54:02,729 pid=89792 INFO - Merging output files
2016-03-30 14:54:02,739 pid=89792 INFO - Removing temporary files
2016-03-30 14:54:02,740 pid=89792 INFO - Done
Required Dependencies:
Optional Dependencies (to generate single file binaries):
Building TACO on your system:
Source files can be obtained from
download section. Unpack the release file (.tar.gz or
.zip) into source directory taco-X.Y.Z. For example:
$ tar -zxvf taco-X.Y.Z.tar.gz
If pyinstaller is installed, the build.sh bash script can be used to compile, install, and generate executable binary files. (note: it might be necessay to chmod +x build.sh to enable the build file to be executed).
e.g.:
$ cd taco-<RELEASE>
$ ./build.sh
Two binary files called taco_run and taco_refcomp are generated in the unpacked directory that can be executed to run TACO and TACO-refcomp.
To install TACO and run without creating an executable, run the following:
$ cd taco-<RELEASE>
$ python setup.py build
The TACO code and reference comparison script can be found in
<install_dir>/build/lib*/taco/ as taco_run.py and taco_refcomp.py. You must either run these scripts from the <install_dir>/build/lib*/taco/ directory:
$ cd build/lib*/taco
$ python taco_run.py -h
$ python taco_refcomp.py -h
or add the build directory to your python path to call this script elsewhere:
$ export PYTHONPATH=<insatll_dir>/build/lib*/taco:$PYTHONPATH
Run TACO from the command line as follows (from either the downloaded platform-specific binary or building from the source code):
$ ./taco_run <options> <gtf_files.txt>
A detailed description of the parameters used to control TACO is below:
<gtf_files.txt>
This required input to TACO is a text file containing paths to files in the
GTF 2.2 format, one per line, produced by an ab initio assembler. For example, the text file passed to TACO could look like this:
/path/to/my/assemblies/mcf7.gtf
/path/to/my/assemblies/bt474.gtf
/path/to/my/assemblies/hela.gtf
General Options
-h/--help
Prints the help message and exits
-v/--verbose
Enables detailed logging for debugging purposes
-p/--num-processes N (default=1)
Run TACO in parallel mode with N processes
-o/--output-dir DIR
Directory where output files will be stored.
This directory must not already exist.
--gtf-expr-attr ATTR (default=FPKM)
GTF attribute field containing expression estimate. The default setting is
FPKM for Cufflinks GTF input.
--filter-min-length N (default 200bp)
Pre-filters input transfrags with length < N prior to assembly. Set to 0 to disable this filter.
--filter-min-expr X (default 0.5)
Pre-filters input transfrags with expression < X. The units of the expression cutoff value X correspond to the units specified by the gtf-expr-attr parameter, which is FPKM by default. Set
to 0.0 to disable this filter.
--isoform-frac FRAC (default 0.05)
Report transcript isoforms with expression fraction >=FRAC relative to the highest expressed gene. For each gene, the highest abundance isoform will be reported with a FRAC of 1.0.
Advanced Options
TACO has several advanced options that can be used to configure specific aspects of its behavior, such as change point detection and splicing pattern network construction. These parameters have been chosen to optimize performance for human
transcriptomes. We recommend that users leave these options at their default settings.
TACO writes output to the directory specified by the
-o command line option. Within this directory, the
import output files are:
Transcriptome assembly: assembly.gtf
This GTF file contains TACO's assembled isoforms. The first 7
columns are standard GTF, and the last column contains attributes,
some of which are also standardized (“gene_id”, and “transcript_id”).
There one GTF record per row, and each record represents either a
transcript or an exon within a transcript. The columns are defined as
follows:
| Column number |
Column name |
Example |
Description |
| 1 |
seqname |
chrX |
Chromosome or contig name |
| 2 |
source |
taco |
The name of the program that generated this file (always taco) |
| 3 |
feature |
exon |
The type of record (always either “transcript” or “exon”. |
| 4 |
start |
77696957 |
The leftmost coordinate of this record (where 1 is the leftmost possible coordinate) |
| 5 |
end |
77712009 |
The rightmost coordinate of this record, inclusive. |
| 6 |
score |
77712009 |
The most abundant isoform for each gene is assigned a score
of 1000. Minor isoforms are scored by the ratio
(minor FPKM/major FPKM) |
| 7 |
strand |
+ |
TACO's guess for which strand the isoform came from.
Always one of “+”, “-“, “.” |
| 7 |
frame |
. |
TACO does not predict where the start and stop
codons (if any) are located within each transcript,
so this field is not used. |
| 8 |
attributes |
… |
See below. |
Each GTF record is decorated with the following attributes:
| Attribute |
Example |
Description |
| gene_id |
G7 |
TACO gene id |
| transcript_id |
TU56 |
TACO transcript id |
| locus_id |
L1 |
TACO locus id |
| tss_id |
TSS31 |
TACO transcription start site id |
| expr |
2.441 |
Isoform-level abundance. The units correspond to the
expression units of the input transfrags (usually FPKM or TPM)
|
| rel_frac |
0.7647 |
Relative abundance of isoform compared to the major isoform
in the gene. The most abundant isoform for each gene is
assigned a rel_frac of 1.0. Minor isoforms are scored
by the ratio (minor expr/major expr)
|
| abs_frac |
0.7647 |
Relative abundance of isoform compared to the total
expression of all isoforms in the gene. Isoforms are scored by
the ratio (expr / sum(expr(x) for each isoform x)).
|
Transcriptome assembly: assembly.bed
This BED file contains TACO's assembled isoforms. Please refer to
the
UCSC genome browser's detailed description of the BED
format. The name column (Column 4) contains
a string of the format gene_id|transcript_id(expr),
e.g. G7|TU56(2.77).
Transfrag coverage profiles:
expr.pos.bedgraph,
expr.neg.bedgraph,
expr.none.bedgraph
TACO outputs 3 bedGraph files with the coverage profile of the
input transfrags on the forward, reverse, and unknown/unspecified
strands. Please refer to the
UCSC genome browser's detailed description of the bedGraph
format. These files can be converted to
bigWig format
using the free conversion tool bedGraphToBigWig for
viewing on genome browsers such as IGV or UCSC.
Transfrag splice junction profiles:
splice_junctions.bed
A UCSC
BED track of junctions reported by TACO. Each junction consists
of two connected BED blocks. The score is the sum of the expression
values of transfrags supporting the junction. This file can be converted
to bigBed
track format for viewing on genome browsers such as IGV or UCSC.
This tool can take any assembly in GTF format and compare to a reference,
and provide details on overlap with reference transcripts,
nature of that overlap, and can also run the CPAT tool to determine coding potential.
We recommend using the GENCODE reference, which can be found
here.
Run reference comparison tool as follows:
$ ./taco_refcomp -o <output_directory> -r <reference_gtf> -t <test_gtf> --cpat (optional flag to run coding potential prediction)
General Options
-h/--help
Prints the help message and exits
-v/--verbose
Enables detailed logging for debugging purposes
-p/--num-processes N (default=1)
Run TACO-refcomp in parallel mode with N processes.
Note that parallelization done at chromosome level.
-r REF
Path to reference gtf to be used in comparison."
-t TEST
Path to gtf being tested (e.g., a TACO produced meta-assembly)."
-o/--output-dir DIR
Directory where output files will be stored.
--cpat (default=false)
Run CPAT tool for coding potential scoring. CPAT function
only supported for human, mouse, and zebrafish. The CPAT tool
can take over an hour depending on the size of the transcriptome
being analyzed.
--cpat-species (human, mouse, zebrafish) (default=human)
Only needed if using --cpat flag.
Species information needed to use approrpriate reference for CPAT algorithm
--cpat-genome GENOME
Only needed if using --cpat flag.
Path to genome fasta file (GENOME) that CPAT utilizes to generate sequence
from assembly. (Genome must be same as that used to generate assemblies)
Note: If --cpat
flag is used, CPAT(v1.2.2) will be run. Please cite the CPAT tool
(reference).
Q. Can you compare a TACO generated assembly to an annotated reference??
A. We provide a reference comparison tool (taco_refcomp) that can be found bundled
with the taco tool. This tool can be used to compare any transcriptome to a reference. The tool also
supports running the CPAT coding potential tool. If CPAT is used, please cite the
CPAT manuscript.
Q. How much memory does TACO require?
A. TACO memory usage scales with the size of the input and the number of parallel processes. Specifically, TACO partitions the input transfrags into independent loci, and transfrags within each loci are stored in memory. Running
TACO with multiple processes allows it to assemble multiple loci in parallel at the expense of additional memory overhead. To examine peak memory usage, we tested TACO on >10,000 datsets. Some of the largest loci contained over 10 million
transfrags each, and memory per process ranged from 3-10GB.
Q. How much time does TACO take to run?
A. TACO runtime also scales with the size of the input and the number of parallel processes. We can offer several examples: 1) TACO processed 55 breast cancer cell line datasets using a Mac OSX laptop computer with 4 cores
in about 10 minutes, and 2) TACO processed ~1000 datasets from the CCLE on a 64-core Linux workstation in about 2 hours and 30 minutes.
Q. Why do I receive a WARNING message that my sort command does not support the --parallel flag?
A. TACO uses the operating system's command line sort to concatenate and sort the input GTF files before beginning assembly. For large jobs (~1000 samples), sorting the GTF can consume a significant portion of
total runtime. Newer versions of GNU coreutils provide a parallelized version of sort. TACO checks to see if sort can be parallelized and defaults to single-threaded sort if this is not supported.
If using TACO, please cite Niknafs et al., Nature Methods, 2017.
Niknafs, Y. S., Pandian, B., Iyer, H. K., Chinnaiyan, A. M., & Iyer, M. K. (2017).
TACO produces robust multisample transcriptome assemblies from RNA-seq. Nat Meth, 14(1), 68–70.
Cufflinks ab initio assemblies of the RNA-Seq datasets from the Cancer Cell Line Encyclopedia (CCLE) are provided for user download. RNA-Seq data were aligned using
STAR prior to running Cufflinks.
Run instructions
First, untar/uncompress the downloaded dataset file:
tar -zxvf ccle_cufflinks.tar.gz
Files will uncompress into a directory e.g. ccle_cufflinks. Within this directory is the file list_of_files.txt which is a list relative paths to the GTF files.
Change your directory to uncompressed directory:
cd ccle_cufflinks
Run TACO with the following command:
./taco_run --gtf-expr-attr FPKM -o my_output_dir list_of_files.txt
Copyright (c) 2015-2018 Matthew Iyer, Yashar Niknafs, Balaji Pandian,
Chinnaiyan Lab, University of Michigan Medical School.
Permission is hereby granted, free of charge, to any person obtaining a copy
of this software and associated documentation files (the "Software"), to deal
in the Software without restriction, including without limitation the rights
to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
copies of the Software, and to permit persons to whom the Software is
furnished to do so, subject to the following conditions:
The above copyright notice and this permission notice shall be included in
all copies or substantial portions of the Software.
THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR
IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY,
FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE
AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER
LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM,
OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN
THE SOFTWARE.
© 2017 Michigan Center for Translational Pathology, University of Michigan Health System